Decoding Gastric Cancer: An AI-Driven Transcriptomic Meta-Analysis
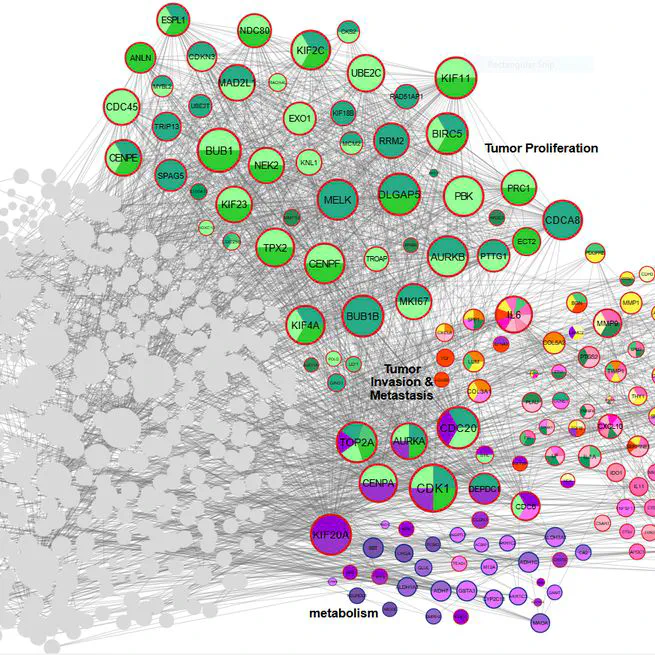
Gastric cancer (GC) remains a leading cause of global cancer mortality, necessitating deeper insights into its molecular mechanisms. This meta-analysis and systematic review integrated transcriptomic data from 28 studies (14 RNA-seq, 13 microarray) to identify critical genes and pathways driving GC progression. Leveraging AI-driven approaches for data harmonization and batch effect correction, we standardized raw datasets from public repositories (GEO, SRA, TCGA) and performed rigorous quality control. Differential expression analysis using edgeR and LIMMA identified 1,163 differentially expressed genes (DEGs), including CST1 (most up-regulated) and PGA3 (most down-regulated). Pathway enrichment revealed tumor proliferation (E2F targets, G2-M checkpoint), ECM remodeling (collagens, MMPs), immune evasion (CXCL chemokines), and metabolic reprogramming as key processes. Protein-protein interaction (PPI) network analysis highlighted hub genes such as AURKA, COL1A1, and IL6, while AI-enhanced clustering delineated functional modules linked to metastasis and prognosis. Survival and immune infiltration analyses underscored the clinical relevance of identified genes. Notably, ERBB4 down-regulation and collagen family up-regulation were mechanistically tied to apoptosis resistance and microenvironment stiffening. AI algorithms further aided in resolving dataset heterogeneity and prioritizing high-confidence biomarkers. This study provides a comprehensive transcriptomic landscape of GC, emphasizing the interplay between genetic drivers, tumor microenvironment, and immune evasion. The integration of AI methodologies enhanced robustness in cross-study data synthesis, offering novel therapeutic targets and underscoring the potential of computational strategies in advancing GC research. These findings illuminate pathways for precision oncology and underscore the need for multi-omics approaches to unravel GC complexity.
Apr 9, 2025
A comprehensive transcriptomic meta-Analysis leveraging deep learning to uncover molecular signatures and potential therapeutic targets in Triple-Negative Breast Cancers
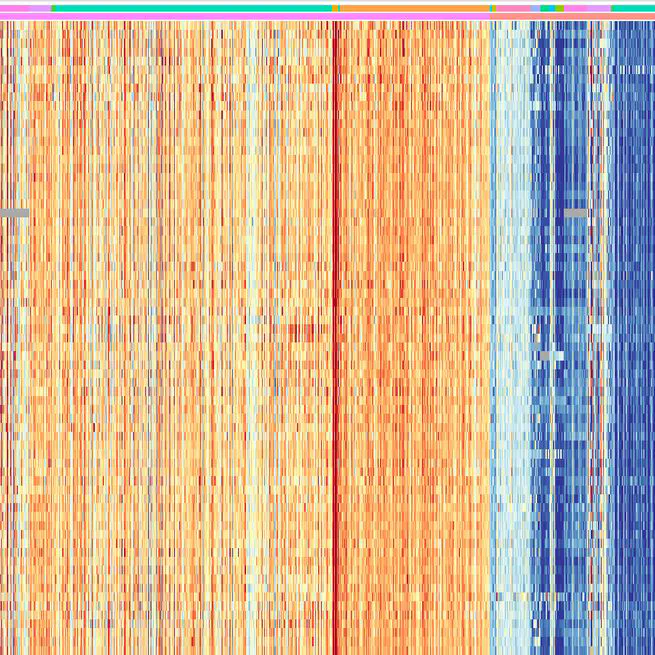
Triple-negative breast cancer (TNBC), characterized by its aggressive behavior and lack of hormone receptor expression, remains a therapeutic challenge. This study integrates multi-omics data and AI-driven approaches to dissect the molecular mechanisms driving TNBC progression. Through a meta-analysis of 49 transcriptomic studies (2013–2024), we identified 2,101 differentially expressed genes (DEGs), including 68 consistently dysregulated protein-coding genes, with CXCL10 (↑4.01-fold) and ADH1B (↓4.8-fold) as the most significantly altered. Pathway enrichment revealed upregulated genes associated with cell proliferation, immune evasion, and metabolic reprogramming, while downregulated genes implicated hormonal signaling suppression and extracellular matrix remodeling. Gene Ontology analysis highlighted mitotic regulation and immune dysregulation as central processes. AI-based clustering of protein-protein interaction networks identified five functional modules (Tumor Growth, Invasion & Metastasis, Metabolism, Immune & Inflammation, Hormonal & Stress Response), with hub genes like CDK1 and CXCL8 driving tumor proliferation and immune escape. Notably, machine learning algorithms enhanced data integration and cluster identification, revealing FOXM1 as a key regulator of mitotic pathways (p = 6.189E-07) and JUN as a mediator of stromal-epithelial interactions despite its downregulation.
Apr 9, 2025
Oleuropein's Effects on Breast Cancer Revealed by RNA-Sequencing and Machine Learning
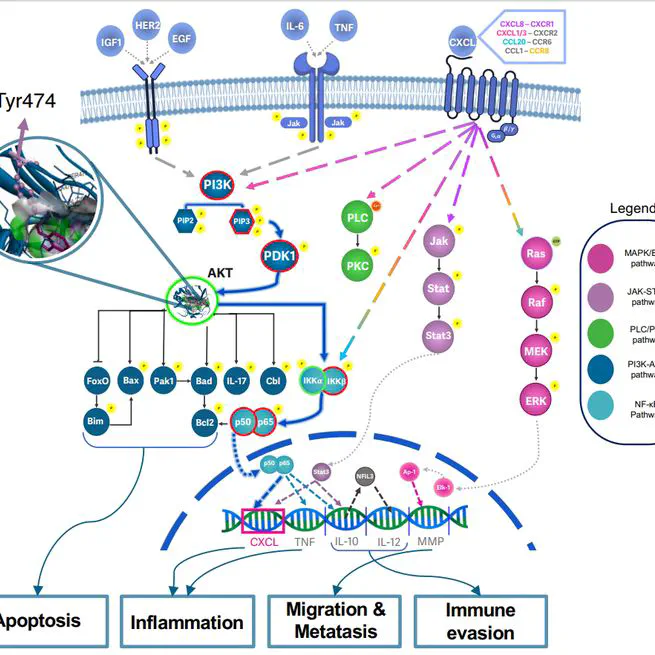
Breast cancer (BC) remains a leading cause of cancer-related morbidity and mortality worldwide, highlighting the Critical need for innovative treatment strategies. Phytochemicals, bioactive compounds derived from plants, have emerged as promising candidates in cancer therapy due to their diverse anti-cancer properties. Oleuropein, a polyphenol found in olive oil, has shown potential in modulating key signaling pathways, inducing apoptosis, and inhibiting metastasis in various cancer models. In this study, we investigated the effects of oleuropein on genome expression profile of MDA-MB-231 BC cell line by RNA-sequencing method. The cell line treated with 200 μL of oleuropein for 48 hours, total RNA extracted from both treated and untreated cells and RNA sequencing performed to assess global gene expression changes. Differential Gene Expression (DEG) analysis was conducted to evaluate pharmacological effects of Oleuropein treatment through pathway analysis and deep learning models. A comprehensive RNA-sequencing analysis revealed a total of 137 differentially expressed genes in MDA-MB-231cells treated with oleuropein. Of these, 115 genes were downregulated, while 21 genes were upregulated during the study. These findings suggest that oleuropein exerts a significant impact on breast cancer cells by modulating multiple molecular mechanisms. The downregulation of numerous genes involved in cell proliferation, survival, and invasion pathways indicates the potential for oleuropein to inhibit tumor growth and metastasis in BC.
Apr 8, 2025

Meta-analysis of transcriptome reveals key genes relating to oil quality in olive
A deep search of RNA-seq published data shed light on thirty-nine experiments associated with the olive transcriptome, four of these proved to be ideal for meta-analysis. Meta-analysis confirmed the genes identified in previous studies and released new genes, which were not identified before. According to the IDR index, the meta-analysis had good power to identify new differentially expressed genes. The key genes were investigated in the metabolic pathways and were grouped into four classes based on the biosynthetic cycle of fatty acids and factors that affect oil quality. Galactose metabolism, glycolysis pathway, pyruvate metabolism, fatty acid biosynthesis, glycerolipid metabolism, and terpenoid backbone biosynthesis were the main pathways in olive oil quality. In galactose metabolism, raffinose is a suitable source of carbon along with other available sources for carbon in fruit development. The results showed that the biosynthesis of acetyl-CoA in glycolysis and pyruvate metabolism is a stable pathway to begin the biosynthesis of fatty acids. Key genes in oleic acid production as an indicator of oil quality and critical genes that played an important role in production of triacylglycerols were identified in different developmental stages. In the minor compound, the terpenoid backbone biosynthesis was investigated and important enzymes were identified as an interconnected network that produces important precursors for the synthesis of a monoterpene, diterpene, triterpene, tetraterpene, and sesquiterpene biosynthesis.
Aug 22, 2023