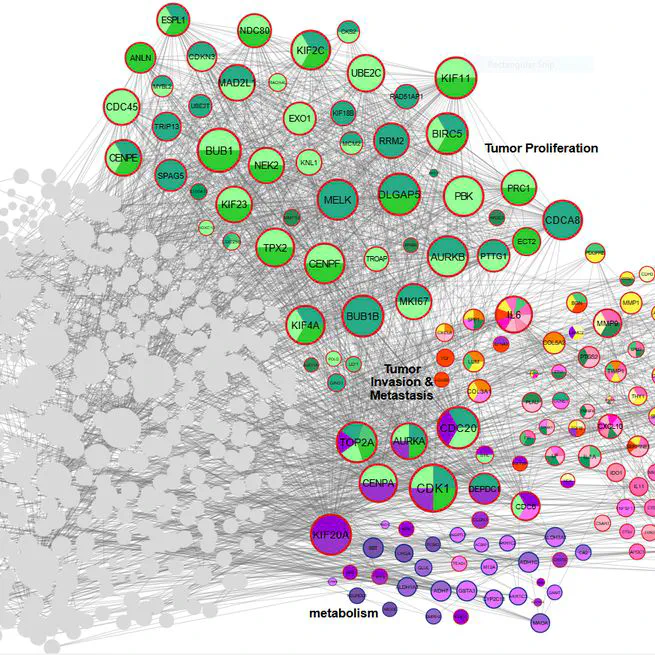
Decoding Gastric Cancer: An AI-Driven Transcriptomic Meta-Analysis
Gastric cancer (GC) remains a leading cause of global cancer mortality, necessitating deeper insights into its molecular mechanisms. This meta-analysis and systematic review integrated transcriptomic data from 28 studies (14 RNA-seq, 13 microarray) to identify critical genes and pathways driving GC progression. Leveraging AI-driven approaches for data harmonization and batch effect correction, we standardized raw datasets from public repositories (GEO, SRA, TCGA) and performed rigorous quality control. Differential expression analysis using edgeR and LIMMA identified 1,163 differentially expressed genes (DEGs), including CST1 (most up-regulated) and PGA3 (most down-regulated). Pathway enrichment revealed tumor proliferation (E2F targets, G2-M checkpoint), ECM remodeling (collagens, MMPs), immune evasion (CXCL chemokines), and metabolic reprogramming as key processes. Protein-protein interaction (PPI) network analysis highlighted hub genes such as AURKA, COL1A1, and IL6, while AI-enhanced clustering delineated functional modules linked to metastasis and prognosis. Survival and immune infiltration analyses underscored the clinical relevance of identified genes. Notably, ERBB4 down-regulation and collagen family up-regulation were mechanistically tied to apoptosis resistance and microenvironment stiffening. AI algorithms further aided in resolving dataset heterogeneity and prioritizing high-confidence biomarkers. This study provides a comprehensive transcriptomic landscape of GC, emphasizing the interplay between genetic drivers, tumor microenvironment, and immune evasion. The integration of AI methodologies enhanced robustness in cross-study data synthesis, offering novel therapeutic targets and underscoring the potential of computational strategies in advancing GC research. These findings illuminate pathways for precision oncology and underscore the need for multi-omics approaches to unravel GC complexity.
Apr 9, 2025
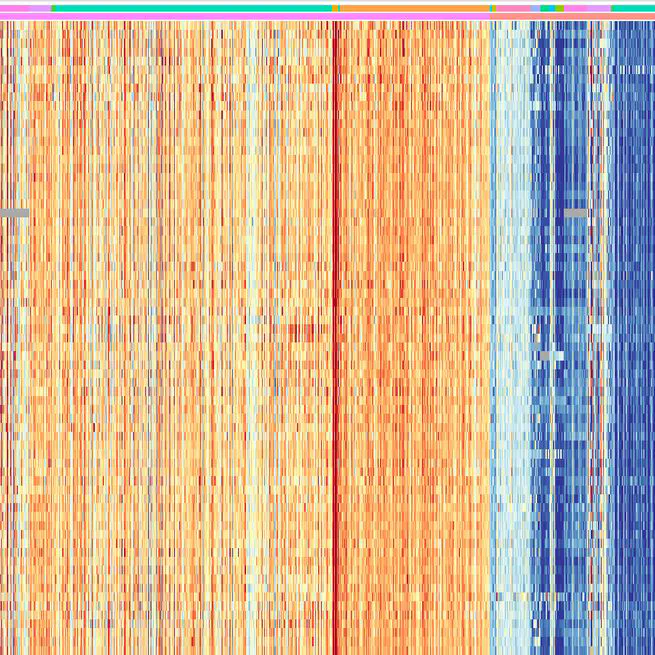
A comprehensive transcriptomic meta-Analysis leveraging deep learning to uncover molecular signatures and potential therapeutic targets in Triple-Negative Breast Cancers
Triple-negative breast cancer (TNBC), characterized by its aggressive behavior and lack of hormone receptor expression, remains a therapeutic challenge. This study integrates multi-omics data and AI-driven approaches to dissect the molecular mechanisms driving TNBC progression. Through a meta-analysis of 49 transcriptomic studies (2013–2024), we identified 2,101 differentially expressed genes (DEGs), including 68 consistently dysregulated protein-coding genes, with CXCL10 (↑4.01-fold) and ADH1B (↓4.8-fold) as the most significantly altered. Pathway enrichment revealed upregulated genes associated with cell proliferation, immune evasion, and metabolic reprogramming, while downregulated genes implicated hormonal signaling suppression and extracellular matrix remodeling. Gene Ontology analysis highlighted mitotic regulation and immune dysregulation as central processes. AI-based clustering of protein-protein interaction networks identified five functional modules (Tumor Growth, Invasion & Metastasis, Metabolism, Immune & Inflammation, Hormonal & Stress Response), with hub genes like CDK1 and CXCL8 driving tumor proliferation and immune escape. Notably, machine learning algorithms enhanced data integration and cluster identification, revealing FOXM1 as a key regulator of mitotic pathways (p = 6.189E-07) and JUN as a mediator of stromal-epithelial interactions despite its downregulation.
Apr 9, 2025
Oleuropein's Effects on Breast Cancer Revealed by RNA-Sequencing and Machine Learning
Breast cancer (BC) remains a leading cause of cancer-related morbidity and mortality worldwide, highlighting the Critical need for innovative treatment strategies. Phytochemicals, bioactive compounds derived from plants, have emerged as promising candidates in cancer therapy due to their diverse anti-cancer properties. Oleuropein, a polyphenol found in olive oil, has shown potential in modulating key signaling pathways, inducing apoptosis, and inhibiting metastasis in various cancer models. In this study, we investigated the effects of oleuropein on genome expression profile of MDA-MB-231 BC cell line by RNA-sequencing method. The cell line treated with 200 μL of oleuropein for 48 hours, total RNA extracted from both treated and untreated cells and RNA sequencing performed to assess global gene expression changes. Differential Gene Expression (DEG) analysis was conducted to evaluate pharmacological effects of Oleuropein treatment through pathway analysis and deep learning models. A comprehensive RNA-sequencing analysis revealed a total of 137 differentially expressed genes in MDA-MB-231cells treated with oleuropein. Of these, 115 genes were downregulated, while 21 genes were upregulated during the study. These findings suggest that oleuropein exerts a significant impact on breast cancer cells by modulating multiple molecular mechanisms. The downregulation of numerous genes involved in cell proliferation, survival, and invasion pathways indicates the potential for oleuropein to inhibit tumor growth and metastasis in BC.
Apr 8, 2025
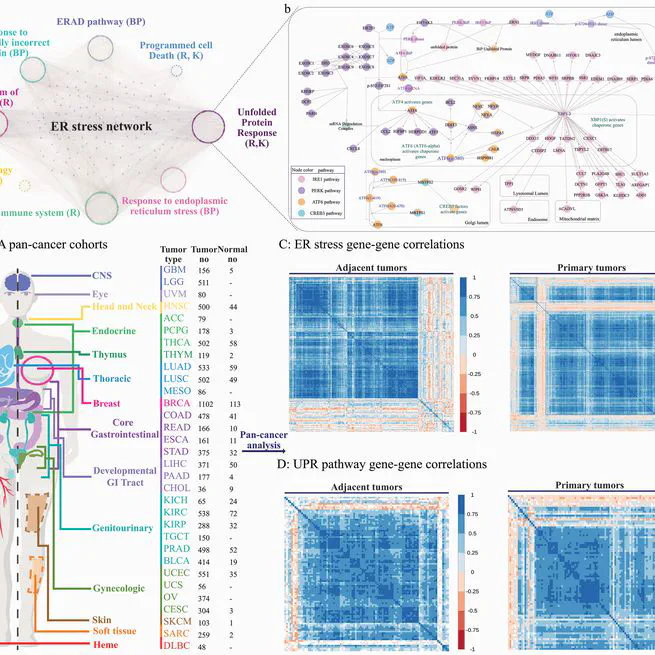
Towards a pan-cancer atlas of endoplasmic reticulum stress network
Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) pathway play pivotal roles in cancer progression and therapy resistance, yet their pan-cancer dynamics and clinical implications remain poorly understood. This study presents a comprehensive analysis of ER stress and UPR pathway activity across 32 cancer types using The Cancer Genome Atlas (TCGA) data. By integrating gene-centric and pathway-centric approaches, including single-sample Gene Set Enrichment Analysis (ssGSEA), we characterized the expression landscape, tumor microenvironment interactions, and clinical relevance of UPR signaling. Our results revealed coordinated ER stress gene expression patterns in primary tumors, with UPR pathway activity significantly elevated in most cancers compared to adjacent normal tissues. Tumor purity inversely correlated with ER stress activity, underscoring microenvironmental influences. Differential expression analysis identified 61 UPR-related genes dysregulated across cancers, with IRE1 and PERK branches predominantly upregulated. Clinically, elevated UPR activity correlated with poor prognosis, advanced tumor stages, and resistance to therapies targeting EGFR, chromatin remodeling, and DNA repair. Co-expression networks highlighted UPR interactions with DNA repair and extracellular matrix pathways, while hallmark pathway analysis linked UPR to mTORC1 signaling, hypoxia, and epithelial-mesenchymal transition. Immune profiling revealed UPR-associated shifts in cytotoxic T cells and macrophages, suggesting microenvironmental modulation. Drug response analysis demonstrated UPR-mediated resistance to EGFR inhibitors and PARP inhibitors, implicating IRE1 as a key contributor. This study establishes the UPR as a central regulator of cancer progression, offering insights into its dual roles in tumor survival and therapy resistance. Our findings advocate for UPR pathway inhibition as a promising strategy to enhance treatment efficacy, particularly in lung, gastrointestinal, and kidney cancers.
Apr 7, 2025
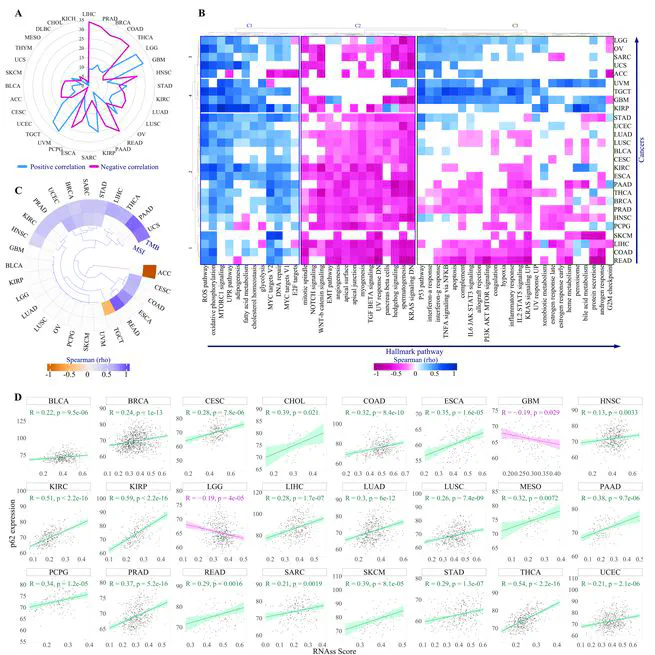
Pan-cancer analysis of SQSTM1/p62 reveals its comprehensive contribution to shaping tumor microenvironment and anti-tumor immunity
Sequestosome 1 (SQSTM1)/p62 is a multifunctional protein involved in diverse physiological processes, and it has been evidenced that its dysregulation implicated in tumorigenesis. Using TCGA pan-cancer data, we analyzed p62 genomic alterations, expression patterns, and clinical relevance. Our results show that p62 mutations and copy number variations (CNVs) are rare, suggesting that prognostic significance of this gene is poor. However, p62 gene expression was significantly elevated in several cancers, including BRCA and LUAD , where it correlated with poorer overall survival and advanced tumor stages. Pathway analyses showed a strong association between p62 and oncogenic features, such as oxidative phosphorylation, reactive oxygen species (ROS), increased tumor mutation burden (TMB), and microsatellite instability (MSI). Intrestingly, p62 expression was inversely associated with immune cell infiltration and positively correlated with immunosuppressive markers, suggesting its role in fostering an immunosuppressive tumor microenvironment (TME) in most types of cancer. Therefore, p62 plays a pivotal role in cancer as both a driver of oncogenesis and a modulator of the TME, supporting its potential as a biomarker and therapeutic target to enhance the efficacy of immunotherapies, particularly immune checkpoint inhibitors (ICIs). Through docking-based virtual screening, we finally identified four natural-product-derived inhibitors targeting the PB1 domain of p62, which is essential for its self-oligomerization, with favorable pharmacokinetic profiles.
Mar 1, 2025
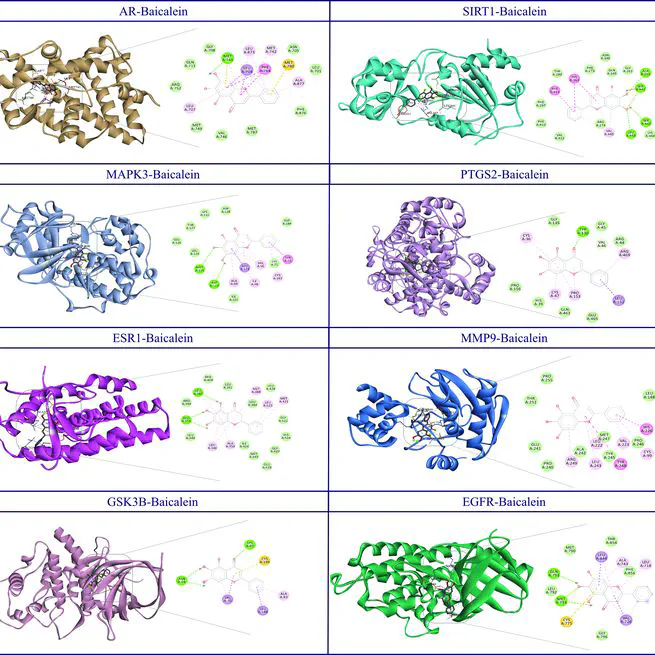
Advanced Molecular Mechanisms of Baicalein's Neuroprotective Effects in Neurodegenerative Disease Treatments
Baicalein is a flavonoid that demonstrates extensive and significant therapeutic potential for numerous neurodegenerative diseases (NDs). Due to its shared influences on the remediation of NDs, our study adopts a comprehensive approach to illuminate the underlying mechanisms responsible for the effects of baicalein. We initiated our investigation by computationally identifying the potential protein targets of baicalein using SwissTargetPrediction in Homo sapiens. Concurrently, we used the DisGeNET database to identify genes related to NDs. By integrating with baicalein-predicted targets, we build an inclusive network that highlights complex relationships between genes, diseases, and baicalein. Our findings revealed that baicalein predominantly affects the AGE/RAGE pathway, interleukin (notably IL-18 and IL-17), and NF-κB signalings, the pathways associated with inflammation and immune-related functions. Furthermore, the effects of baicalein extend to the pathways with critical roles in NDs, such as BDNF and PI3K-Akt signaling. Using protein-protein interaction networks to validate our findings, we identified key hub proteins (AR, EGFR, SIRT1, MAPK3, APP, ESR1, PTGS2, MMP9, and GSK3B) that may mediate the therapeutic effects of baicalein against NDs. In this case, molecular docking indicates strong binding affinities between them and baicalein. In summary, our detailed study highlights baicalein's potential as a promising treatment for NDs, offering a molecular basis for its effectiveness.
Feb 24, 2025
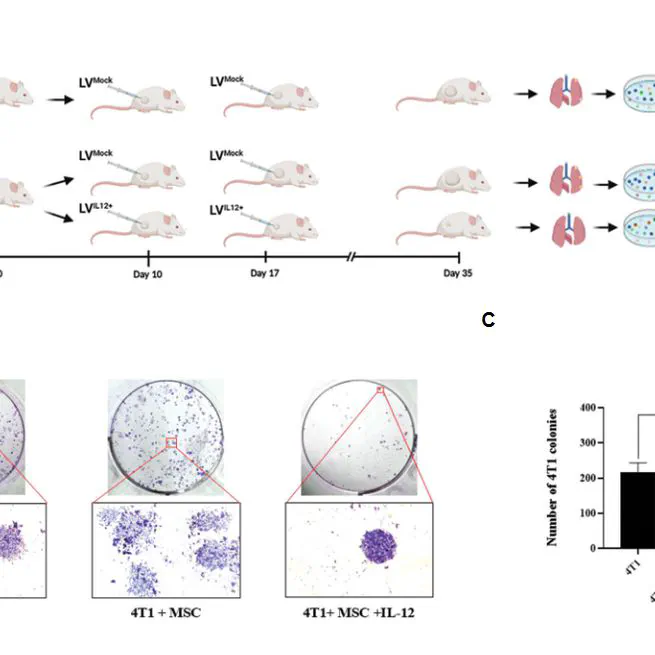
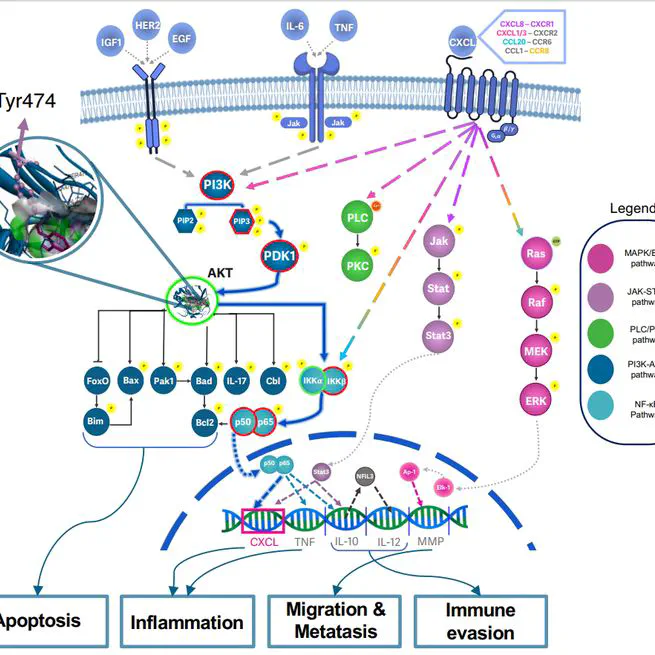
Interleukin-12 Inhibits Tumor Growth and Metastasis Promoted by Tumor-Associated Mesenchymal Stem Cells in Triple-Negative Breast Cancer
The aim of this study was to understand the interactions between tumor-associated mesenchymal stem cells (TA-MSCs) and triple-negative breast cancer (TNBC) cells, which appear to be necessary for developing effective therapies. The findings of the present study revealed a complex interplay between TA-MSCs and TNBC cells that affects tumor growth and metastasis. Preclinical results indicate that intratumoral IL-12 immunotherapy shows promise in overcoming TA-MSC-promoted tumor growth and metastasis.
Jan 11, 2025
Cancers Meta-analysis
The Cancers Meta-analysis Projects integrate comprehensive transcriptome, genome, and epigenome datasets to advance precision oncology through rigorous meta-analysis and cutting-edge AI models. Focused on high-impact cancers—including gastric, triple-negative breast cancer (TNBC), colorectal, lung, glioblastoma, and pancreatic cancers.
Jan 26, 2024
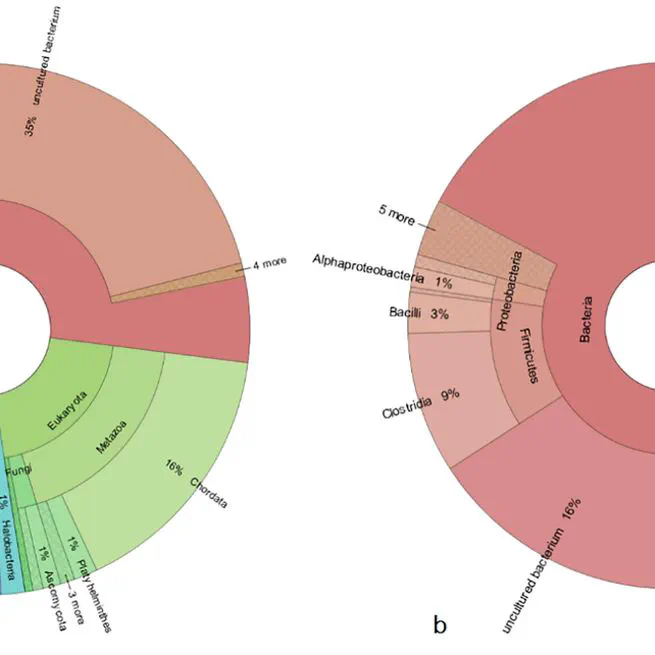
Genomic palaeoparasitology traced the occurrence of Taenia asiatica in ancient Iran (Sassanid Empire, 2th cent. CE–6th cent. CE)
Palaeoparasitology investigates parasitological infections in animals and humans of past distance by examining biological remains. Palaeofaeces (or coprolites) are biological remains that provide valuable information on the disease, diet, and population movements in ancient times. Today, advances in detecting ancient DNA have cast light on dark corners that microscopy could never reach. The archaeological site of the Chehrabad salt mine of Achaemenid (550–330 BC) and Sassanid (third–seventh century AD) provides remains of various biotic and abiotic samples, including animal coprolites, for multidisciplinary studies. In the present work, we investigated coprolites for helminth eggs and larvae by microscopy and traced their biological agents’ DNA by Next Generation Sequencing. Our results revealed various helminths, including Taenia asiatica, the species introduced in the 1990s. Implementing advanced modern molecular techniques like NGS gives a paramount view of pathogenic agents in space and time.
Jul 14, 2022
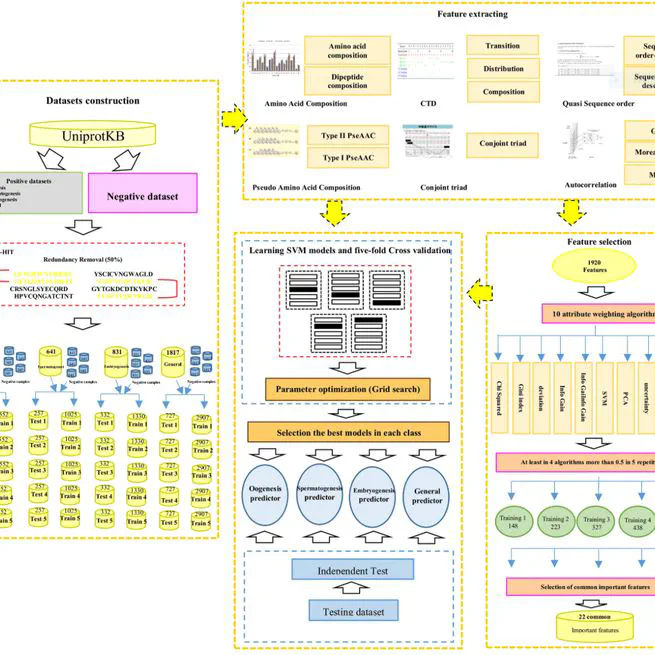
PrESOgenesis: a two-layer multi-label predictor for identifying fertility-related proteins using support vector machine and pseudo amino acid composition approach
Successful spermatogenesis and oogenesis are the two genetically independent processes preceding embryo development. To date, several fertility-related proteins have been described in mammalian species. Nevertheless, further studies are required to discover more proteins associated with the development of germ cells and embryogenesis in order to shed more light on the processes. This work builds on our previous software (OOgenesis_Pred), mainly focusing on algorithms beyond what was previously done, in particular new fertility-related proteins and their classes (embryogenesis, spermatogenesis and oogenesis) based on the support vector machine according to the concept of Chou’s pseudo-amino acid composition features. The results of five-fold cross validation, as well as the independent test demonstrated that this method is capable of predicting the fertility-related proteins and their classes with accuracy of more than 80%. Moreover, by using feature selection methods, important properties of fertility-related proteins were identified that allowed for their accurate classification. Based on the proposed method, a two-layer classifier software, named as “PrESOgenesis” (https://github.com/mrb20045/PrESOgenesis) was developed. The tool identified a query sequence (protein or transcript) as fertility or non-fertility-related protein at the first layer and then classified the predicted fertility-related protein into different classes of embryogenesis, spermatogenesis or oogenesis at the second layer.
Jun 13, 2018