About
- Bioinformatics
- Genomics
- Artificial Intelligence
- Computational biology
- Systems biology
PhD in Genomics
Sant' Anna University, Italy, 2010
MSc in Molecular Biology
University of Shiraz, Iran, 2007
BSc in Molecular Biology
University of Mazandaran, Iran, 2004
Experience
Senior Bioinformatician
NIGEB- Population genome diversity
- Pan-cancer studies
- Meta-analysis, and omics integration
- Drug discovery
- AI models
- Train and lead junior scientists
Scientific Director of Genome Center
NIGEB- Genomics services and lab establishment
- Analysis pipelines development
- Scientific consults
Bioinformatics Research Scientist
NIGEB- Pan and core genome analysis
- Algorithms for the identification and annotation of genetic variants
- Bioinformatics support for research projects
- Data mining, comparative genomics
- Transcriptome Analysis
- Population genetics
Research Assistant
University of Milan- Analysis of genomic data
- Partial chromosome assembly for research project
- Statistical analysis and visualization
- Preparation of publications and research reports
Education
PhD in Genomics
Sant' Anna University, Italy, 2010Top studentMSc in Molecular Biology
University of Shiraz, Iran, 2007GPA: 3.8/4.0BSc in Molecular Biology
University of Mazandaran, Iran, 2004GPA: 3.4/4.0
Data Analysis, Pipeline Development, Backend (Flask/Django)
95%
Statistical Analysis, Visualization
80%
Scripting, NGS Pipeline Automation, System Management
95%
Full-Stack Development (React, Node.js, REST APIs)
80%
Database Design & Querying
70%
Genomics, Transcriptomics, Proteomics
90%
Network Analysis, Pathway Modeling
90%
Supervised/Unsupervised Models, CNNs, GNNs, VAE, LLM
80%
Pan-genome, GWAS, MetaQTL, Demographic Analysis
75%
Drug-Target Prediction, Virtual Screening
75%
Analytical, Innovative, Adaptable
95%
Technical Writing, Presentations, Stakeholder Collaboration
95%
Cross-functional Collaboration
95%
Self-directed Research, Project Ownership
95%
Continuous proactive engagement with emerging fields
95%
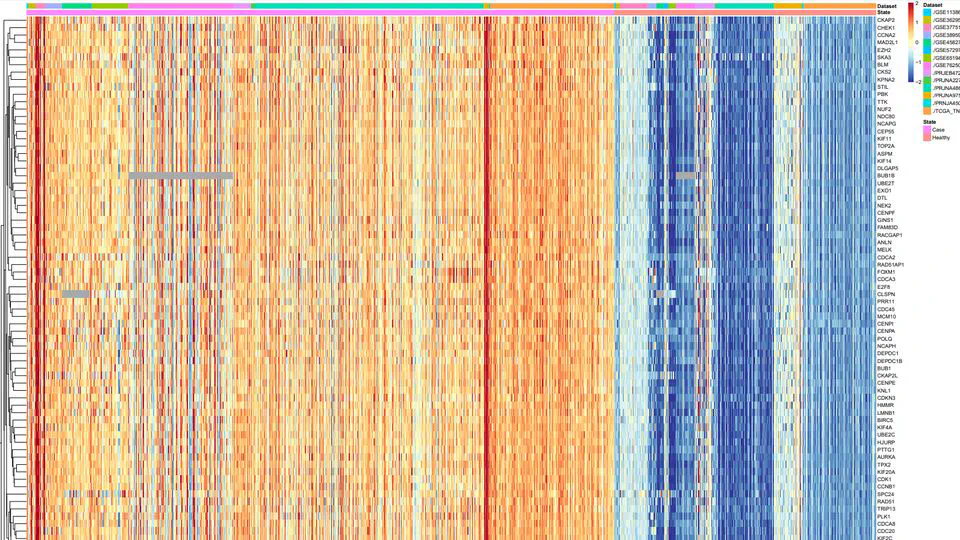
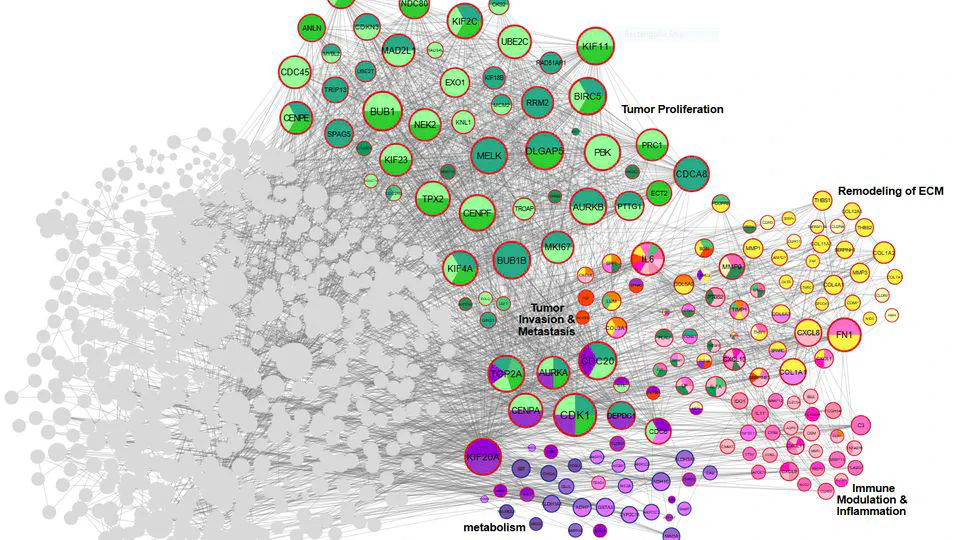
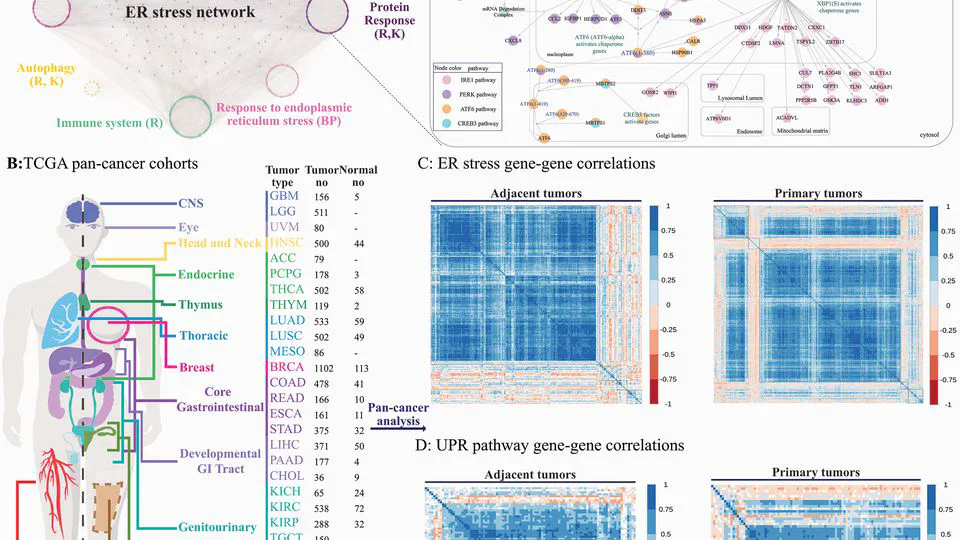
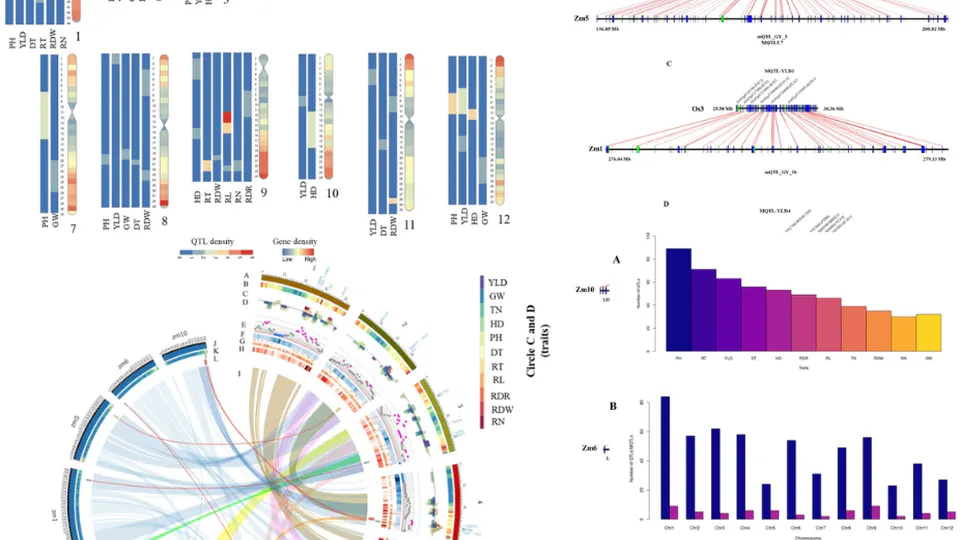
The Cancers Meta-analysis Projects integrate comprehensive transcriptome, genome, and epigenome datasets to advance precision oncology through rigorous meta-analysis and cutting-edge AI models. Focused on high-impact cancers—including gastric, triple-negative breast cancer (TNBC), colorectal, lung, glioblastoma, and pancreatic cancers.
Jan 26, 2024

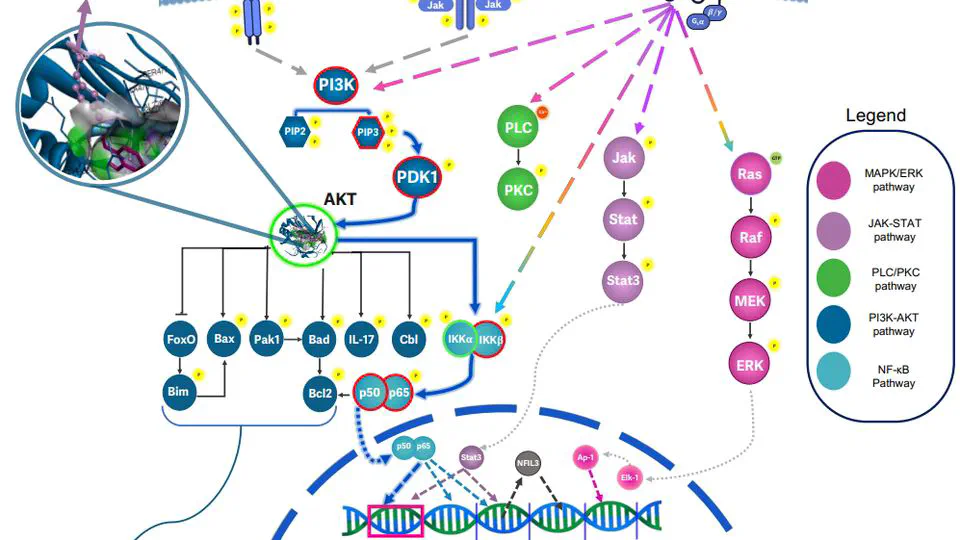
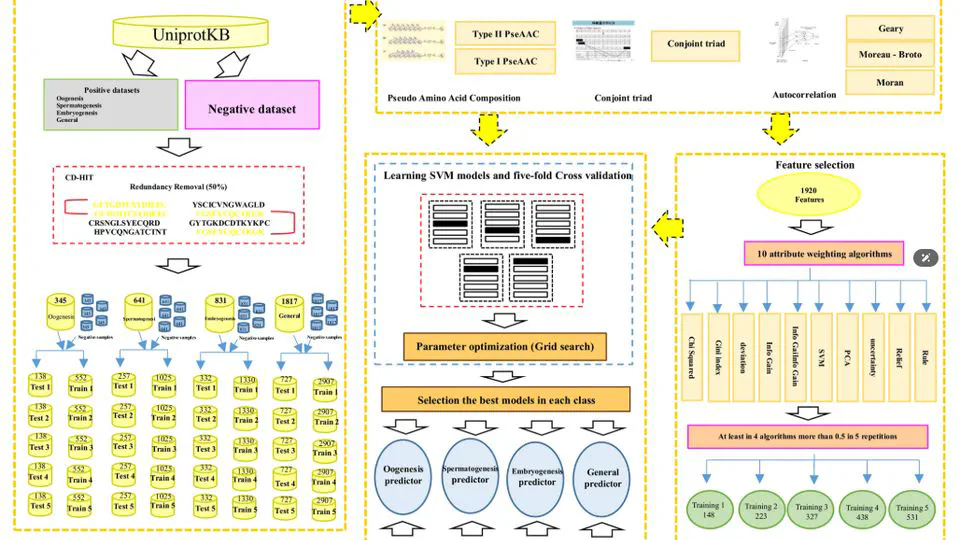
Projects bridges AI-driven target prioritization with precision experimental pipelines to uncover novel therapeutics. Leveraging structural bioinformatics and high-throughput virtual screening, the platform computationally evaluates millions of compounds against metagenes—AI-identified genes critical to disease mechanisms (e.g., cancer progression, antimicrobial resistance).
Jan 13, 2024

Delivering modular, software solutions to streamline data analysis, diagnostics, and industrial workflows across research and commercial sectors. By developing customizable pipelines in Python (automation, machine learning integration), Bash (high-performance computing orchestration), R (statistical modeling, visualization), and JavaScript (interactive web interfaces), the projects empowers users to process complex datasets with reproducibility and scalability.
Nov 21, 2023

Project harnesses global genome and transcriptome sequencing of olive genotypes to decode biodiversity, evolutionary adaptation, and human-driven migration patterns. By assembling high-resolution datasets from wild and cultivated varieties across continents, the project maps genetic diversity hotspots, identifies loci underpinning traits like drought tolerance and fruit yield, and reconstructs ancient dispersal routes shaped by trade and agriculture.
Oct 26, 2023
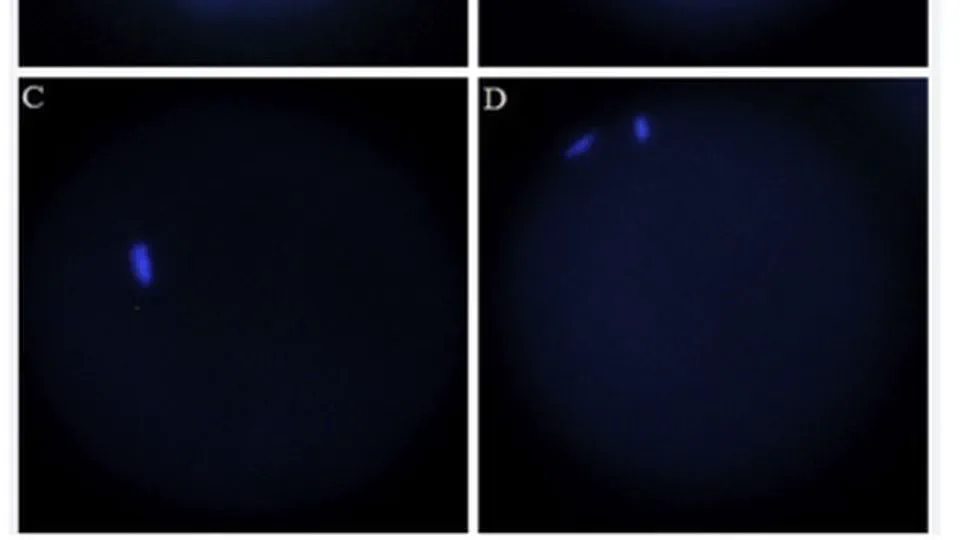
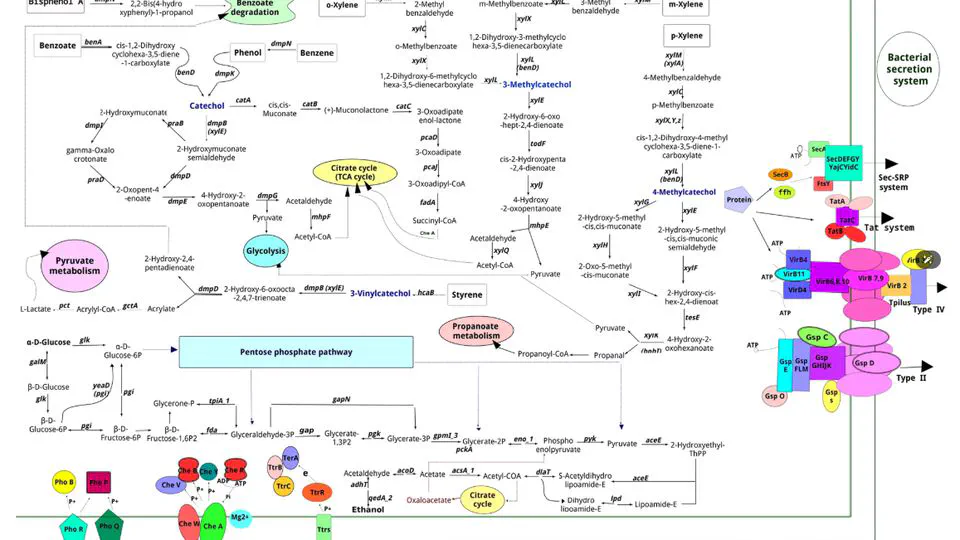
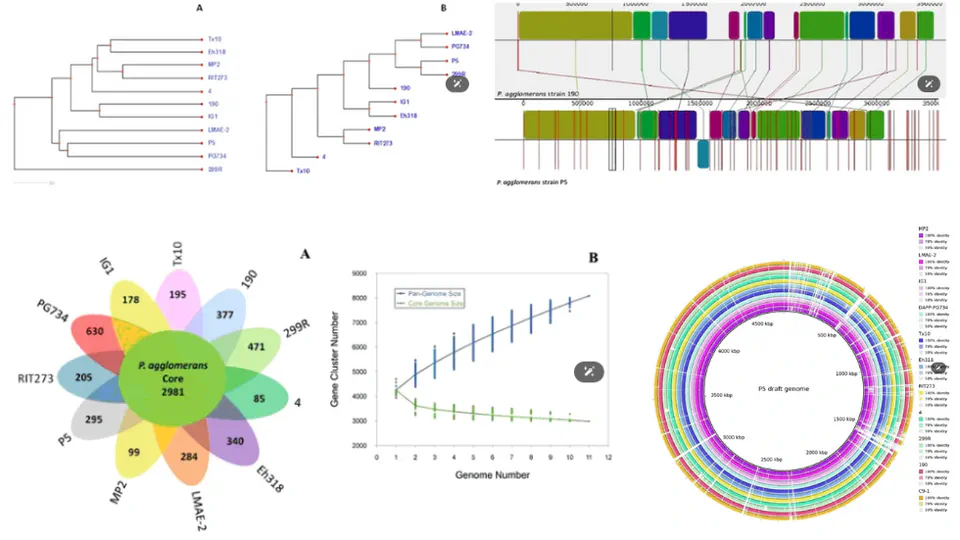
leveraging high-resolution genome sequencing and annotation to decode the functional and evolutionary landscapes of bacteria, fungi, and viruses. By systematically assembling and annotating small genomes across diverse taxa and ecological niches, the project uncovers conserved metabolic pathways, virulence factors, and niche-specific adaptations.
Oct 22, 2023