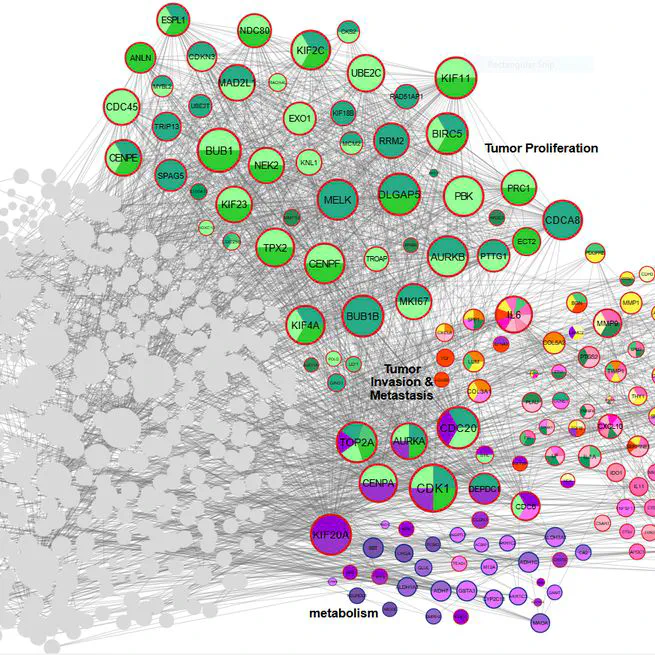
Decoding Gastric Cancer: An AI-Driven Transcriptomic Meta-Analysis
Gastric cancer (GC) remains a leading cause of global cancer mortality, necessitating deeper insights into its molecular mechanisms. This meta-analysis and systematic review integrated transcriptomic data from 28 studies (14 RNA-seq, 13 microarray) to identify critical genes and pathways driving GC progression. Leveraging AI-driven approaches for data harmonization and batch effect correction, we standardized raw datasets from public repositories (GEO, SRA, TCGA) and performed rigorous quality control. Differential expression analysis using edgeR and LIMMA identified 1,163 differentially expressed genes (DEGs), including CST1 (most up-regulated) and PGA3 (most down-regulated). Pathway enrichment revealed tumor proliferation (E2F targets, G2-M checkpoint), ECM remodeling (collagens, MMPs), immune evasion (CXCL chemokines), and metabolic reprogramming as key processes. Protein-protein interaction (PPI) network analysis highlighted hub genes such as AURKA, COL1A1, and IL6, while AI-enhanced clustering delineated functional modules linked to metastasis and prognosis. Survival and immune infiltration analyses underscored the clinical relevance of identified genes. Notably, ERBB4 down-regulation and collagen family up-regulation were mechanistically tied to apoptosis resistance and microenvironment stiffening. AI algorithms further aided in resolving dataset heterogeneity and prioritizing high-confidence biomarkers. This study provides a comprehensive transcriptomic landscape of GC, emphasizing the interplay between genetic drivers, tumor microenvironment, and immune evasion. The integration of AI methodologies enhanced robustness in cross-study data synthesis, offering novel therapeutic targets and underscoring the potential of computational strategies in advancing GC research. These findings illuminate pathways for precision oncology and underscore the need for multi-omics approaches to unravel GC complexity.
Apr 9, 2025
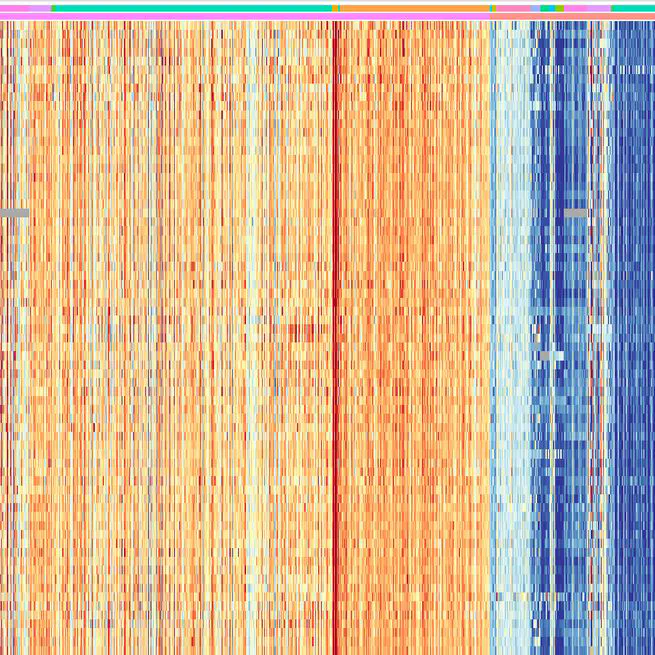
A comprehensive transcriptomic meta-Analysis leveraging deep learning to uncover molecular signatures and potential therapeutic targets in Triple-Negative Breast Cancers
Triple-negative breast cancer (TNBC), characterized by its aggressive behavior and lack of hormone receptor expression, remains a therapeutic challenge. This study integrates multi-omics data and AI-driven approaches to dissect the molecular mechanisms driving TNBC progression. Through a meta-analysis of 49 transcriptomic studies (2013–2024), we identified 2,101 differentially expressed genes (DEGs), including 68 consistently dysregulated protein-coding genes, with CXCL10 (↑4.01-fold) and ADH1B (↓4.8-fold) as the most significantly altered. Pathway enrichment revealed upregulated genes associated with cell proliferation, immune evasion, and metabolic reprogramming, while downregulated genes implicated hormonal signaling suppression and extracellular matrix remodeling. Gene Ontology analysis highlighted mitotic regulation and immune dysregulation as central processes. AI-based clustering of protein-protein interaction networks identified five functional modules (Tumor Growth, Invasion & Metastasis, Metabolism, Immune & Inflammation, Hormonal & Stress Response), with hub genes like CDK1 and CXCL8 driving tumor proliferation and immune escape. Notably, machine learning algorithms enhanced data integration and cluster identification, revealing FOXM1 as a key regulator of mitotic pathways (p = 6.189E-07) and JUN as a mediator of stromal-epithelial interactions despite its downregulation.
Apr 9, 2025
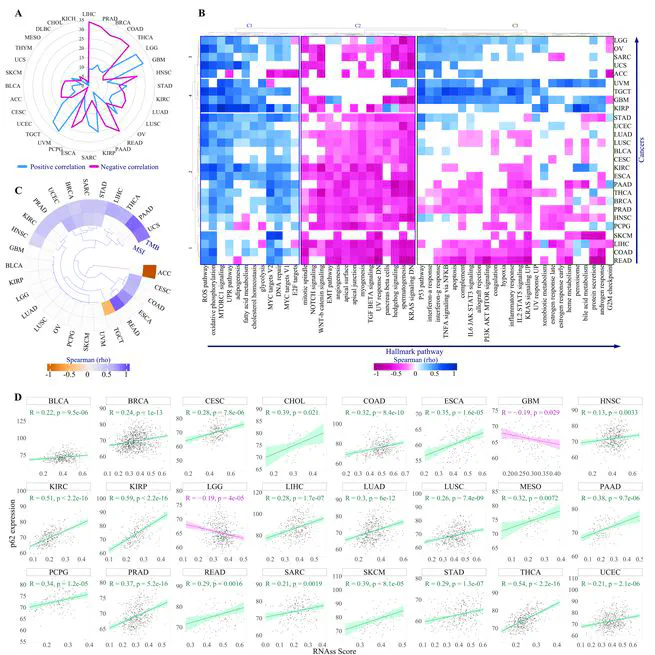
Pan-cancer analysis of SQSTM1/p62 reveals its comprehensive contribution to shaping tumor microenvironment and anti-tumor immunity
Sequestosome 1 (SQSTM1)/p62 is a multifunctional protein involved in diverse physiological processes, and it has been evidenced that its dysregulation implicated in tumorigenesis. Using TCGA pan-cancer data, we analyzed p62 genomic alterations, expression patterns, and clinical relevance. Our results show that p62 mutations and copy number variations (CNVs) are rare, suggesting that prognostic significance of this gene is poor. However, p62 gene expression was significantly elevated in several cancers, including BRCA and LUAD , where it correlated with poorer overall survival and advanced tumor stages. Pathway analyses showed a strong association between p62 and oncogenic features, such as oxidative phosphorylation, reactive oxygen species (ROS), increased tumor mutation burden (TMB), and microsatellite instability (MSI). Intrestingly, p62 expression was inversely associated with immune cell infiltration and positively correlated with immunosuppressive markers, suggesting its role in fostering an immunosuppressive tumor microenvironment (TME) in most types of cancer. Therefore, p62 plays a pivotal role in cancer as both a driver of oncogenesis and a modulator of the TME, supporting its potential as a biomarker and therapeutic target to enhance the efficacy of immunotherapies, particularly immune checkpoint inhibitors (ICIs). Through docking-based virtual screening, we finally identified four natural-product-derived inhibitors targeting the PB1 domain of p62, which is essential for its self-oligomerization, with favorable pharmacokinetic profiles.
Mar 1, 2025

Olive Genomics
Project harnesses global genome and transcriptome sequencing of olive genotypes to decode biodiversity, evolutionary adaptation, and human-driven migration patterns. By assembling high-resolution datasets from wild and cultivated varieties across continents, the project maps genetic diversity hotspots, identifies loci underpinning traits like drought tolerance and fruit yield, and reconstructs ancient dispersal routes shaped by trade and agriculture.
Oct 26, 2023
Small Genomes
leveraging high-resolution genome sequencing and annotation to decode the functional and evolutionary landscapes of bacteria, fungi, and viruses. By systematically assembling and annotating small genomes across diverse taxa and ecological niches, the project uncovers conserved metabolic pathways, virulence factors, and niche-specific adaptations.
Oct 22, 2023
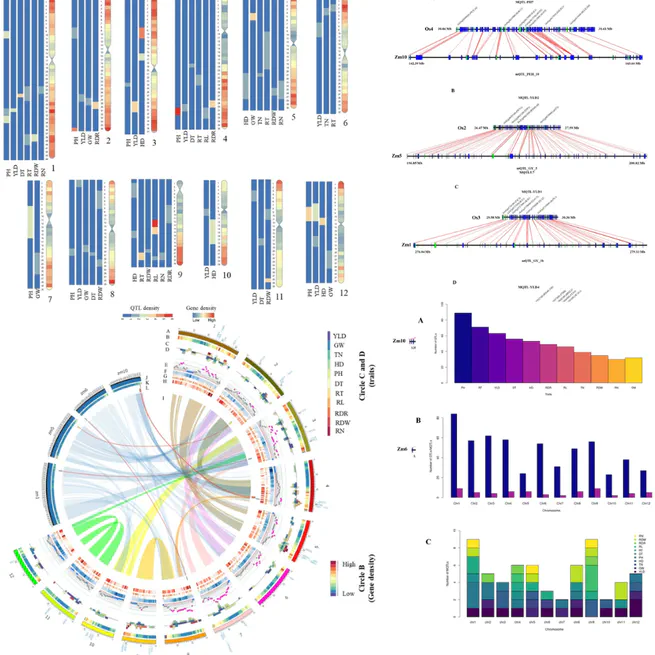
Meta-QTL and ortho-MQTL analyses identified genomic regions controlling rice yield, yield-related traits and root architecture under water deficit conditions
Meta-QTL (MQTL) analysis is a robust approach for genetic dissection of complex quantitative traits. Rice varieties adapted to non-flooded cultivation are highly desirable in breeding programs due to the water deficit global problem. In order to identify stable QTLs for major agronomic traits under water deficit conditions, we performed a comprehensive MQTL analysis on 563 QTLs from 67 rice populations published from 2001 to 2019. Yield and yield-related traits including grain weight, heading date, plant height, tiller number as well as root architecture-related traits including root dry weight, root length, root number, root thickness, the ratio of deep rooting and plant water content under water deficit condition were investigated. A total of 61 stable MQTLs over different genetic backgrounds and environments were identified. The average confidence interval of MQTLs was considerably refined compared to the initial QTLs, resulted in the identification of some well-known functionally characterized genes and several putative novel CGs for investigated traits. Ortho-MQTL mining based on genomic collinearity between rice and maize allowed identification of five ortho-MQTLs between these two cereals. The results can help breeders to improve yield under water deficit conditions.
Mar 25, 2021

Comprehensive genomic analysis of an indigenous Pseudomonas pseudoalcaligenes degrading phenolic compounds
Environmental contamination with aromatic compounds is a universal challenge. Aromatic-degrading microorganisms isolated from the same or similar polluted environments seem to be more suitable for bioremediation. Moreover, microorganisms adapted to contaminated environments are able to use toxic compounds as the sole sources of carbon and energy. An indigenous strain of Pseudomonas, isolated from the Mahshahr Petrochemical plant in the Khuzestan province, southwest of Iran, was studied genetically. It was characterized as a novel Gram-negative, aerobic, halotolerant, rod-shaped bacterium designated Pseudomonas YKJ, which was resistant to chloramphenicol and ampicillin. Genome of the strain was completely sequenced using Illumina technology to identify its genetic characteristics. MLST analysis revealed that the YKJ strain belongs to the genus Pseudomonas indicating the highest sequence similarity with Pseudomonas pseudoalcaligenes strain CECT 5344 (99% identity). Core- and pan-genome analysis indicated that P. pseudoalcaligenes contains 1,671 core and 3,935 unique genes for coding DNA sequences. The metabolic and degradation pathways for aromatic pollutants were investigated using the NCBI and KEGG databases. Genomic and experimental analyses showed that the YKJ strain is able to degrade certain aromatic compounds including bisphenol A, phenol, benzoate, styrene, xylene, benzene and chlorobenzene. Moreover, antibiotic resistance and chemotaxis properties of the YKJ strain were found to be controlled by two-component regulatory systems.
Sep 4, 2019
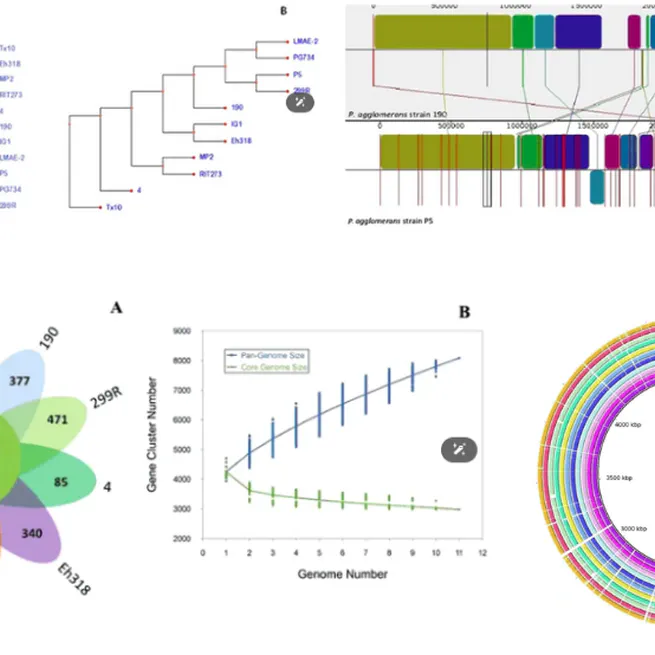
Comprehensive genomic analysis of a plant growth-promoting rhizobacterium Pantoea agglomerans strain P5
In this study, we provide a comparative genomic analysis of Pantoea agglomerans strain P5 and 10 closely related strains based on phylogenetic analyses. A next-generation shotgun strategy was implemented using the Illumina HiSeq 2500 technology followed by core- and pan-genome analysis. The genome of P. agglomerans strain P5 contains an assembly size of 5082485 bp with 55.4% G + C content. P. agglomerans consists of 2981 core and 3159 accessory genes for Coding DNA Sequences (CDSs) based on the pan-genome analysis. Strain P5 can be grouped closely with strains PG734 and 299 R using pan and core genes, respectively. All the predicted and annotated gene sequences were allocated to KEGG pathways. Accordingly, genes involved in plant growth-promoting (PGP) ability, including phosphate solubilization, IAA and siderophore production, acetoin and 2,3-butanediol synthesis and bacterial secretion, were assigned. This study provides an in-depth view of the PGP characteristics of strain P5, highlighting its potential use in agriculture as a biofertilizer.
Nov 15, 2017