Decoding Gastric Cancer: An AI-Driven Transcriptomic Meta-Analysis
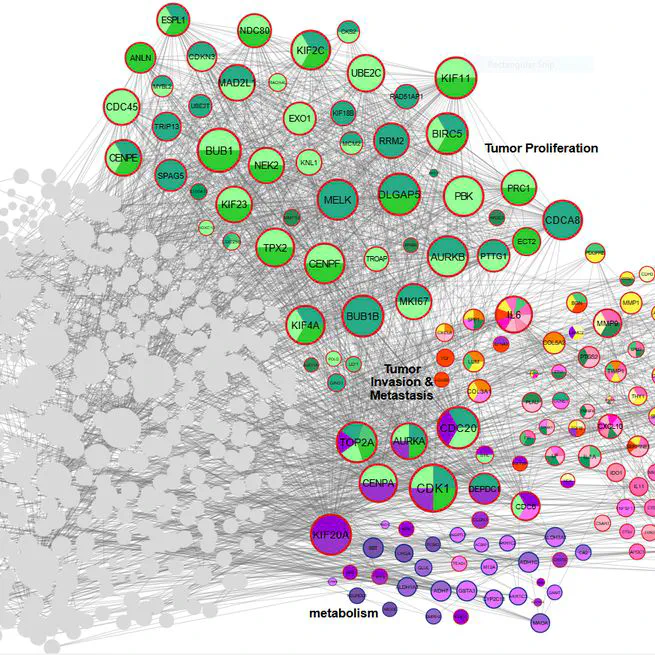
Gastric cancer (GC) remains a leading cause of global cancer mortality, necessitating deeper insights into its molecular mechanisms. This meta-analysis and systematic review integrated transcriptomic data from 28 studies (14 RNA-seq, 13 microarray) to identify critical genes and pathways driving GC progression. Leveraging AI-driven approaches for data harmonization and batch effect correction, we standardized raw datasets from public repositories (GEO, SRA, TCGA) and performed rigorous quality control. Differential expression analysis using edgeR and LIMMA identified 1,163 differentially expressed genes (DEGs), including CST1 (most up-regulated) and PGA3 (most down-regulated). Pathway enrichment revealed tumor proliferation (E2F targets, G2-M checkpoint), ECM remodeling (collagens, MMPs), immune evasion (CXCL chemokines), and metabolic reprogramming as key processes. Protein-protein interaction (PPI) network analysis highlighted hub genes such as AURKA, COL1A1, and IL6, while AI-enhanced clustering delineated functional modules linked to metastasis and prognosis. Survival and immune infiltration analyses underscored the clinical relevance of identified genes. Notably, ERBB4 down-regulation and collagen family up-regulation were mechanistically tied to apoptosis resistance and microenvironment stiffening. AI algorithms further aided in resolving dataset heterogeneity and prioritizing high-confidence biomarkers. This study provides a comprehensive transcriptomic landscape of GC, emphasizing the interplay between genetic drivers, tumor microenvironment, and immune evasion. The integration of AI methodologies enhanced robustness in cross-study data synthesis, offering novel therapeutic targets and underscoring the potential of computational strategies in advancing GC research. These findings illuminate pathways for precision oncology and underscore the need for multi-omics approaches to unravel GC complexity.
Apr 9, 2025
A comprehensive transcriptomic meta-Analysis leveraging deep learning to uncover molecular signatures and potential therapeutic targets in Triple-Negative Breast Cancers
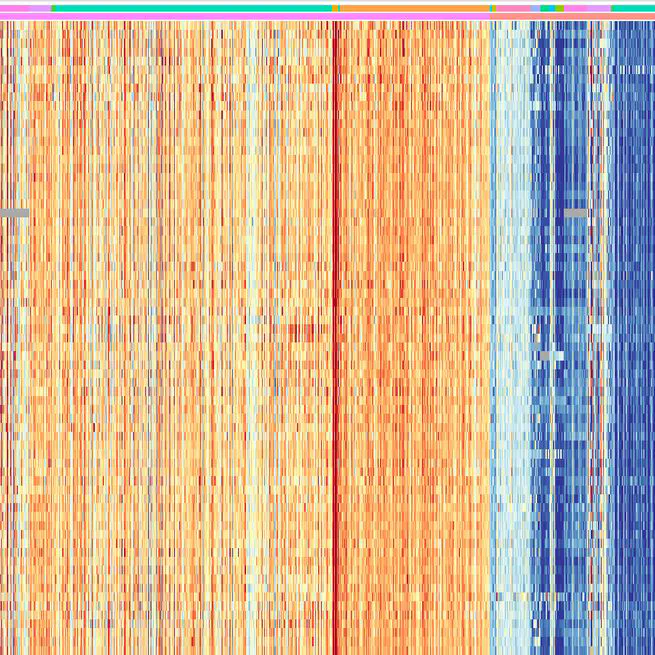
Triple-negative breast cancer (TNBC), characterized by its aggressive behavior and lack of hormone receptor expression, remains a therapeutic challenge. This study integrates multi-omics data and AI-driven approaches to dissect the molecular mechanisms driving TNBC progression. Through a meta-analysis of 49 transcriptomic studies (2013–2024), we identified 2,101 differentially expressed genes (DEGs), including 68 consistently dysregulated protein-coding genes, with CXCL10 (↑4.01-fold) and ADH1B (↓4.8-fold) as the most significantly altered. Pathway enrichment revealed upregulated genes associated with cell proliferation, immune evasion, and metabolic reprogramming, while downregulated genes implicated hormonal signaling suppression and extracellular matrix remodeling. Gene Ontology analysis highlighted mitotic regulation and immune dysregulation as central processes. AI-based clustering of protein-protein interaction networks identified five functional modules (Tumor Growth, Invasion & Metastasis, Metabolism, Immune & Inflammation, Hormonal & Stress Response), with hub genes like CDK1 and CXCL8 driving tumor proliferation and immune escape. Notably, machine learning algorithms enhanced data integration and cluster identification, revealing FOXM1 as a key regulator of mitotic pathways (p = 6.189E-07) and JUN as a mediator of stromal-epithelial interactions despite its downregulation.
Apr 9, 2025
Towards a pan-cancer atlas of endoplasmic reticulum stress network
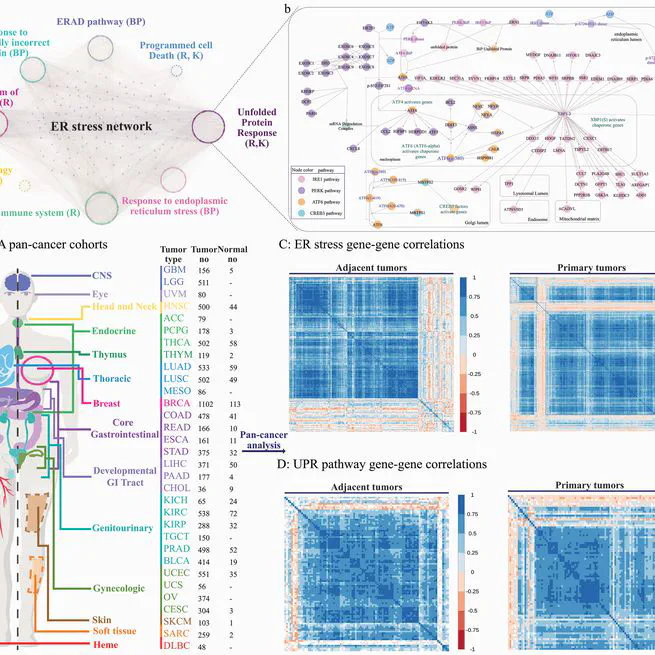
Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) pathway play pivotal roles in cancer progression and therapy resistance, yet their pan-cancer dynamics and clinical implications remain poorly understood. This study presents a comprehensive analysis of ER stress and UPR pathway activity across 32 cancer types using The Cancer Genome Atlas (TCGA) data. By integrating gene-centric and pathway-centric approaches, including single-sample Gene Set Enrichment Analysis (ssGSEA), we characterized the expression landscape, tumor microenvironment interactions, and clinical relevance of UPR signaling. Our results revealed coordinated ER stress gene expression patterns in primary tumors, with UPR pathway activity significantly elevated in most cancers compared to adjacent normal tissues. Tumor purity inversely correlated with ER stress activity, underscoring microenvironmental influences. Differential expression analysis identified 61 UPR-related genes dysregulated across cancers, with IRE1 and PERK branches predominantly upregulated. Clinically, elevated UPR activity correlated with poor prognosis, advanced tumor stages, and resistance to therapies targeting EGFR, chromatin remodeling, and DNA repair. Co-expression networks highlighted UPR interactions with DNA repair and extracellular matrix pathways, while hallmark pathway analysis linked UPR to mTORC1 signaling, hypoxia, and epithelial-mesenchymal transition. Immune profiling revealed UPR-associated shifts in cytotoxic T cells and macrophages, suggesting microenvironmental modulation. Drug response analysis demonstrated UPR-mediated resistance to EGFR inhibitors and PARP inhibitors, implicating IRE1 as a key contributor. This study establishes the UPR as a central regulator of cancer progression, offering insights into its dual roles in tumor survival and therapy resistance. Our findings advocate for UPR pathway inhibition as a promising strategy to enhance treatment efficacy, particularly in lung, gastrointestinal, and kidney cancers.
Apr 7, 2025