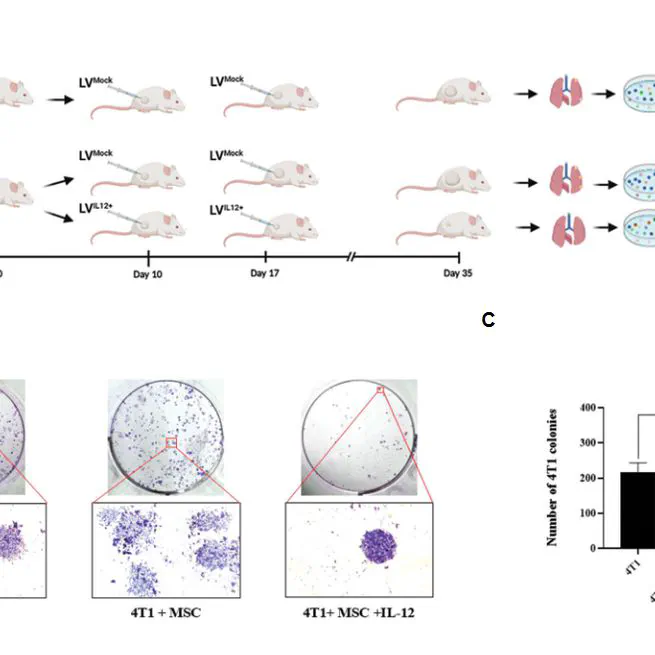
Interleukin-12 Inhibits Tumor Growth and Metastasis Promoted by Tumor-Associated Mesenchymal Stem Cells in Triple-Negative Breast Cancer
The aim of this study was to understand the interactions between tumor-associated mesenchymal stem cells (TA-MSCs) and triple-negative breast cancer (TNBC) cells, which appear to be necessary for developing effective therapies. The findings of the present study revealed a complex interplay between TA-MSCs and TNBC cells that affects tumor growth and metastasis. Preclinical results indicate that intratumoral IL-12 immunotherapy shows promise in overcoming TA-MSC-promoted tumor growth and metastasis.
Jan 11, 2025
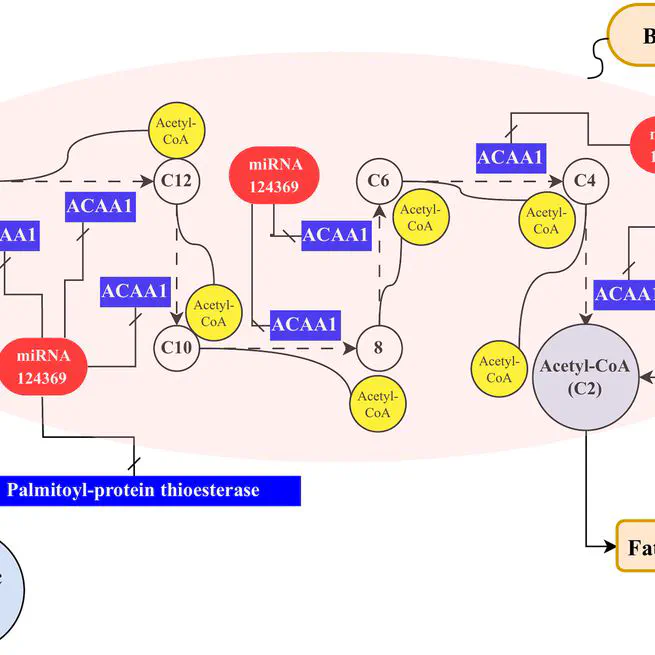
Machine learning-aided microRNA discovery for olive oil quality
MicroRNAs (miRNAs) are key regulators of gene expression in plants, influencing various biological processes such as oil quality and seed development. Although, our knowledge about miRNAs in olive (Olea europaea L.) is progressing, with several miRNAs being identified in previous studies, but most of these reported miRNAs have been predicted without the aid of a reference genome, primarily due to limited genome accessibility at the time. However, significant knowledge gaps still need to be improved in this area. This study addresses the complexities of miRNA detection in olive, using a high quality reference genome and a combination of genomics and machine learning-based methods. By leveraging random forest and support vector machine algorithms, we successfully identified 56 novel miRNAs in olive, surpassing the limitations of conventional homology-based methods. Our subsequent analysis revealed that some of these miRNAs are implicated in the regulation of key genes involved in oil quality. Within the context of oil biosynthesis pathways, the novel miRNA Oeu124369 regulates fatty acid biosynthesis by targeting acetyl-CoA acyltransferase 1 and palmitoyl-protein thioesterase, thereby influencing the production of acetyl-CoA and palmitic acid, respectively. These findings underscore the power of machine learning in unraveling the complex miRNA regulatory network in olive and provide a high quality miRNA resource for future research aimed at improving olive oil production by exploring the target genes of the identified miRNAs to understand their role and their biological processes.
Oct 11, 2024
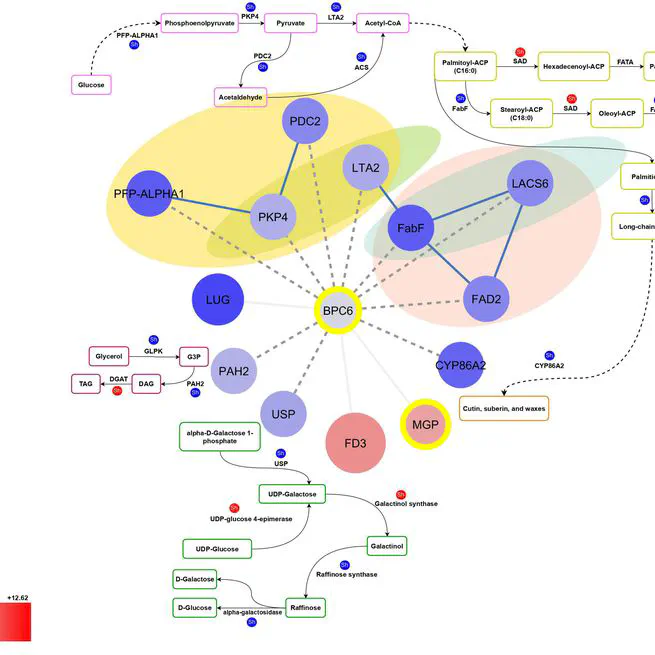
Unraveling the genetic basis of oil quality in olives: a comparative transcriptome analysis
The balanced fatty acid profile of olive oil not only enhances its stability but also contributes to its positive effects on health, making it a valuable dietary choice. Olive oil's high content of unsaturated fatty acids and low content of saturated fatty acids contribute to its beneficial effects on cardiovascular diseases and cancer. The quantities of these fatty acids in olive oil may fluctuate due to various factors, with genotype being a crucial determinant of the oil's quality. This study investigated the genetic basis of oil quality by comparing the transcriptome of two Iranian cultivars with contrasting oil profiles; Mari, known for its high oleic acid content, and Shengeh, characterized by high linoleic acid at Jaén index four. Gas chromatography confirmed a significant difference in fatty acid composition between the two cultivars. Mari exhibited significantly higher oleic acid content (78.48%) compared to Shengeh (48.05%), while linoleic acid content was significantly lower in Mari (4.76%) than in Shengeh (26.69%). Using RNA sequencing at Jaén index four, we analyzed genes involved in fatty acid biosynthesis. Differential expression analysis identified 2775 genes showing statistically significant differences between the cultivars. Investigating these genes across nine fundamental pathways involved in oil quality led to the identification of 25 effective genes. Further analysis revealed 78 transcription factors and 95 transcription binding sites involved in oil quality, with BPC6 and RGA emerging as unique factors. This research provides a comprehensive understanding of the genetic and molecular mechanisms underlying oil quality in olive cultivars. The findings have practical implications for olive breeders and producers, potentially streamlining cultivar selection processes and contributing to the production of high-quality olive oil.
Oct 1, 2024
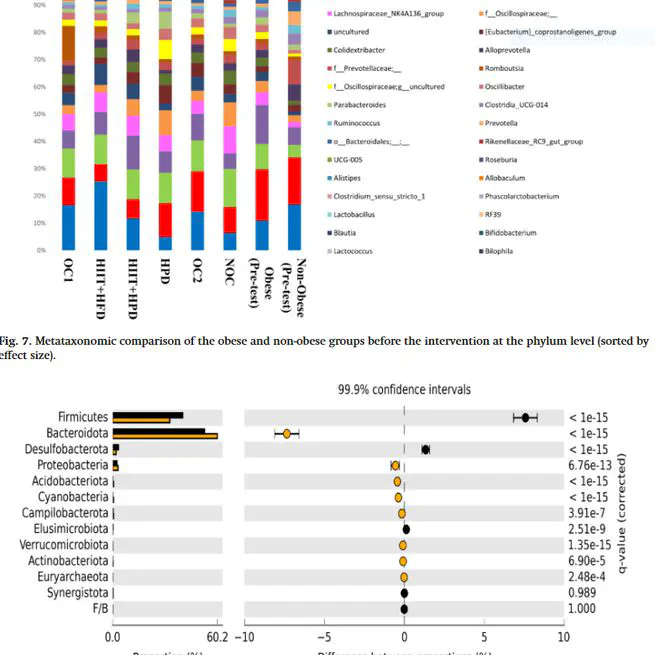
Interaction between high-intensity interval training and high-protein diet on gut microbiota composition and body weight in obese male rats
Diet and exercise are two critical factors that regulate gut microbiota, affecting weight management. The present study investigated the effect of 10 weeks of high-intensity interval training (HIIT) and a high-protein diet (HPD) on gut microbiota composition and body weight changes in obese male Wistar rats. Forty obese rats were randomly divided into five groups, including HPD, HIIT + HPD, HIIT + high-fat diet (HFD) (continuing HFD during intervention), obese control 1 (continuing HFD during intervention), obese control 2 (cutting off HFD at the beginning of the intervention and continuing standard diet), and eight non-obese Wistar rats as a non-obese control (NOC) group (standard diet). Microbial community composition and diversity analysis by sequencing 16S rRNA genes derived from the fecal samples, body weight, and Lee index were assessed. The body weight and Lee index in the NOC, HIIT + HFD, HPD, and HIIT + HPD groups were significantly lower than that in the OC1 and OC2 groups along with the lower body weight and Lee index in the HPD and HIIT + HPD groups compared with the HIIT + HFD group. Also, HFD consumption and switching from HFD to a standard diet or HPD increased gut microbiota dysbiosis. Furthermore, HIIT along with HFD increased the adverse effects of HFD on gut microbiota, while the HIIT + HPD increased microbial richness, improved gut microbiota dysbiosis, and changed rats’ phenotype to lean. It appears that HFD discontinuation without doing HIIT does not improve gut microbiota dysbiosis. Also, the HIIT + HFD, HPD, and HIIT + HPD slow down HFD-induced weight gain, but HIIT + HPD is a more reliable strategy for weight management due to its beneficial effects on gut microbiota composition.
Aug 29, 2023

Meta-analysis of transcriptome reveals key genes relating to oil quality in olive
A deep search of RNA-seq published data shed light on thirty-nine experiments associated with the olive transcriptome, four of these proved to be ideal for meta-analysis. Meta-analysis confirmed the genes identified in previous studies and released new genes, which were not identified before. According to the IDR index, the meta-analysis had good power to identify new differentially expressed genes. The key genes were investigated in the metabolic pathways and were grouped into four classes based on the biosynthetic cycle of fatty acids and factors that affect oil quality. Galactose metabolism, glycolysis pathway, pyruvate metabolism, fatty acid biosynthesis, glycerolipid metabolism, and terpenoid backbone biosynthesis were the main pathways in olive oil quality. In galactose metabolism, raffinose is a suitable source of carbon along with other available sources for carbon in fruit development. The results showed that the biosynthesis of acetyl-CoA in glycolysis and pyruvate metabolism is a stable pathway to begin the biosynthesis of fatty acids. Key genes in oleic acid production as an indicator of oil quality and critical genes that played an important role in production of triacylglycerols were identified in different developmental stages. In the minor compound, the terpenoid backbone biosynthesis was investigated and important enzymes were identified as an interconnected network that produces important precursors for the synthesis of a monoterpene, diterpene, triterpene, tetraterpene, and sesquiterpene biosynthesis.
Aug 22, 2023
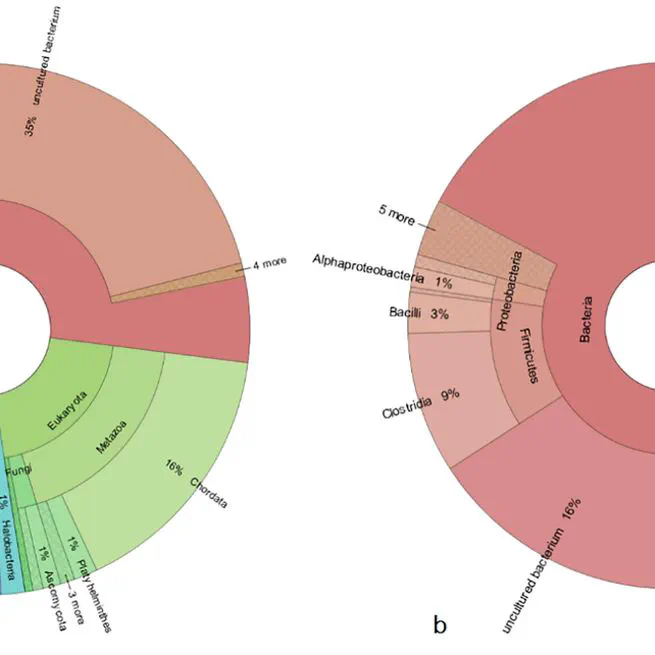
Genomic palaeoparasitology traced the occurrence of Taenia asiatica in ancient Iran (Sassanid Empire, 2th cent. CE–6th cent. CE)
Palaeoparasitology investigates parasitological infections in animals and humans of past distance by examining biological remains. Palaeofaeces (or coprolites) are biological remains that provide valuable information on the disease, diet, and population movements in ancient times. Today, advances in detecting ancient DNA have cast light on dark corners that microscopy could never reach. The archaeological site of the Chehrabad salt mine of Achaemenid (550–330 BC) and Sassanid (third–seventh century AD) provides remains of various biotic and abiotic samples, including animal coprolites, for multidisciplinary studies. In the present work, we investigated coprolites for helminth eggs and larvae by microscopy and traced their biological agents’ DNA by Next Generation Sequencing. Our results revealed various helminths, including Taenia asiatica, the species introduced in the 1990s. Implementing advanced modern molecular techniques like NGS gives a paramount view of pathogenic agents in space and time.
Jul 14, 2022
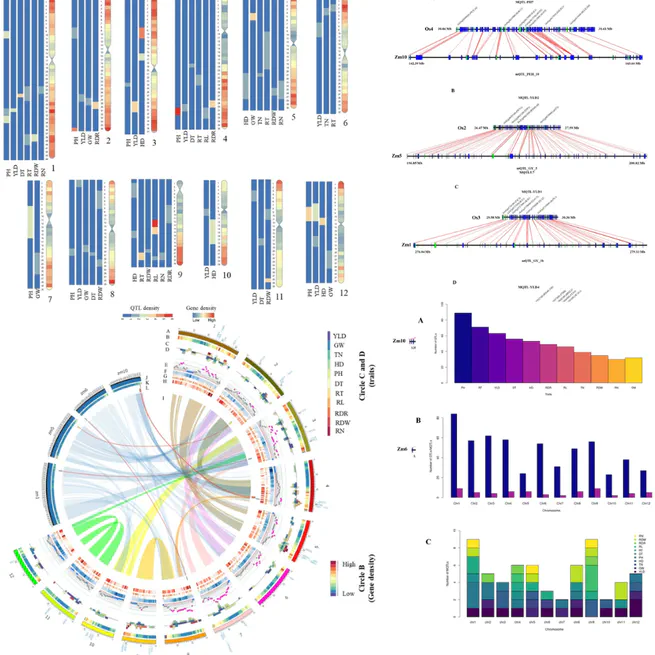
Meta-QTL and ortho-MQTL analyses identified genomic regions controlling rice yield, yield-related traits and root architecture under water deficit conditions
Meta-QTL (MQTL) analysis is a robust approach for genetic dissection of complex quantitative traits. Rice varieties adapted to non-flooded cultivation are highly desirable in breeding programs due to the water deficit global problem. In order to identify stable QTLs for major agronomic traits under water deficit conditions, we performed a comprehensive MQTL analysis on 563 QTLs from 67 rice populations published from 2001 to 2019. Yield and yield-related traits including grain weight, heading date, plant height, tiller number as well as root architecture-related traits including root dry weight, root length, root number, root thickness, the ratio of deep rooting and plant water content under water deficit condition were investigated. A total of 61 stable MQTLs over different genetic backgrounds and environments were identified. The average confidence interval of MQTLs was considerably refined compared to the initial QTLs, resulted in the identification of some well-known functionally characterized genes and several putative novel CGs for investigated traits. Ortho-MQTL mining based on genomic collinearity between rice and maize allowed identification of five ortho-MQTLs between these two cereals. The results can help breeders to improve yield under water deficit conditions.
Mar 25, 2021
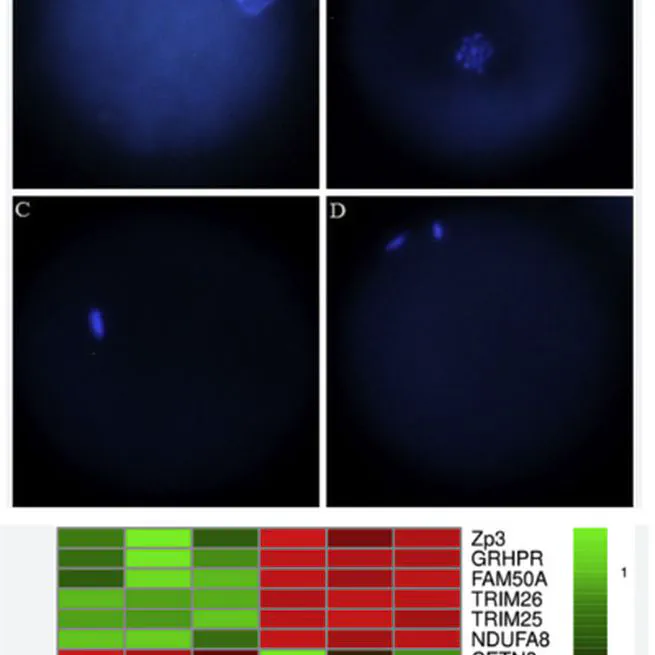
Transcriptional profile of ovine oocytes matured under lipopolysaccharide treatment in vitro
Lipopolysaccharide (LPS) derived from gram negative bacteria cell wall is known to cause ruminal acidosis and/or infectious diseases such as metritis and mastitis which has a significant negative impact on the reproductive performance. This study aimed to investigate the effect of LPS on oocyte maturation and subsequent development in vitro. Ovine cumulus oocyte complexes (COCs) were matured in a medium supplemented with 0 (control), 0.01, 0.1, 1 and 10 μg/mL LPS. Nuclear maturation, cleavage and blastocyst rate, mitochondrial membrane potential (ΔΨm), intracellular reactive oxygen species (ROS) content and changes to the transcript abundance were evaluated. In case of the maturation rate, the percentage of oocytes reaching the MII stage was lower following exposure to 10 μg/mL LPS in comparison to the control group (P < 0.05). Moreover, the blastocyst rate decreased in case of 1 and 10 μg/mL LPS when compared to the control group (P < 0.05). ROS overproduction accompanied by a decreased ΔΨm were recorded in LPS treated oocytes in comparison to the control group (P < 0.05). The 3′ tag digital gene expression profiling method revealed that 7887 genes were expressed while only seven genes exhibited changes in the transcript abundance following exposure to LPS. Tripartite motif containing 25 (TRIM25), Tripartite motif containing 26 (TRIM26), Zona Pellucida glycoprotein 3 (ZP3), Family with sequence similarity 50-member A (FAM50A), Glyoxalate and hydroxy pyruvate reductase (GRHPR), NADH ubiquinase oxireductase subunit A8 (NDUFA8) were down-regulated (P < 0.05), while only Centrin 3 (CETN3) was up-regulated (P < 0.05). Our findings show that LPS has undesirable effects on the maturation competence of ovine oocytes and subsequent embryo development. In addition, the transcriptomic profiling results may shed more light on the molecular mechanisms of LPS-induced infertility in ruminants.
Nov 1, 2020

Comprehensive genomic analysis of an indigenous Pseudomonas pseudoalcaligenes degrading phenolic compounds
Environmental contamination with aromatic compounds is a universal challenge. Aromatic-degrading microorganisms isolated from the same or similar polluted environments seem to be more suitable for bioremediation. Moreover, microorganisms adapted to contaminated environments are able to use toxic compounds as the sole sources of carbon and energy. An indigenous strain of Pseudomonas, isolated from the Mahshahr Petrochemical plant in the Khuzestan province, southwest of Iran, was studied genetically. It was characterized as a novel Gram-negative, aerobic, halotolerant, rod-shaped bacterium designated Pseudomonas YKJ, which was resistant to chloramphenicol and ampicillin. Genome of the strain was completely sequenced using Illumina technology to identify its genetic characteristics. MLST analysis revealed that the YKJ strain belongs to the genus Pseudomonas indicating the highest sequence similarity with Pseudomonas pseudoalcaligenes strain CECT 5344 (99% identity). Core- and pan-genome analysis indicated that P. pseudoalcaligenes contains 1,671 core and 3,935 unique genes for coding DNA sequences. The metabolic and degradation pathways for aromatic pollutants were investigated using the NCBI and KEGG databases. Genomic and experimental analyses showed that the YKJ strain is able to degrade certain aromatic compounds including bisphenol A, phenol, benzoate, styrene, xylene, benzene and chlorobenzene. Moreover, antibiotic resistance and chemotaxis properties of the YKJ strain were found to be controlled by two-component regulatory systems.
Sep 4, 2019
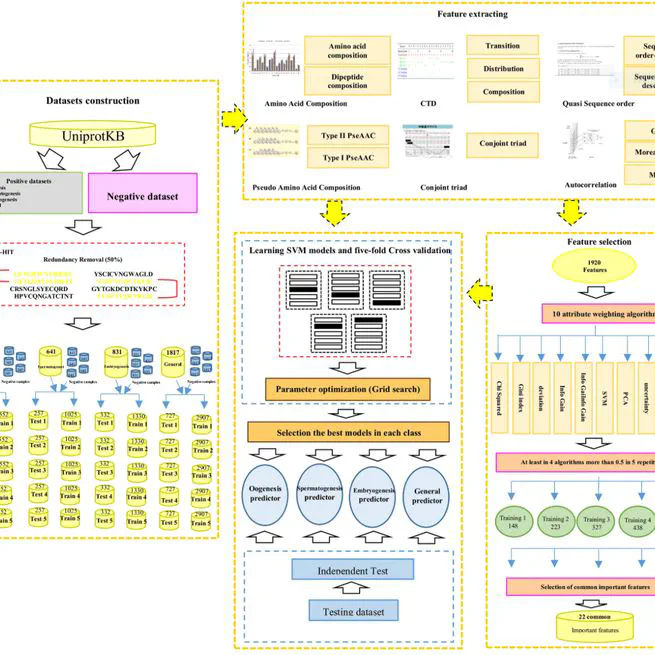
PrESOgenesis: a two-layer multi-label predictor for identifying fertility-related proteins using support vector machine and pseudo amino acid composition approach
Successful spermatogenesis and oogenesis are the two genetically independent processes preceding embryo development. To date, several fertility-related proteins have been described in mammalian species. Nevertheless, further studies are required to discover more proteins associated with the development of germ cells and embryogenesis in order to shed more light on the processes. This work builds on our previous software (OOgenesis_Pred), mainly focusing on algorithms beyond what was previously done, in particular new fertility-related proteins and their classes (embryogenesis, spermatogenesis and oogenesis) based on the support vector machine according to the concept of Chou’s pseudo-amino acid composition features. The results of five-fold cross validation, as well as the independent test demonstrated that this method is capable of predicting the fertility-related proteins and their classes with accuracy of more than 80%. Moreover, by using feature selection methods, important properties of fertility-related proteins were identified that allowed for their accurate classification. Based on the proposed method, a two-layer classifier software, named as “PrESOgenesis” (https://github.com/mrb20045/PrESOgenesis) was developed. The tool identified a query sequence (protein or transcript) as fertility or non-fertility-related protein at the first layer and then classified the predicted fertility-related protein into different classes of embryogenesis, spermatogenesis or oogenesis at the second layer.
Jun 13, 2018